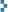
Refining Uranium by the PUREX Process
September 18, 2009 PUREX is the major chemical technique for recovering uranium from spent nuclear fuel. Based on the highly-selective extraction of uranyl nitrate from aqueous solution by tributyl phosphate (TBP) in a nonpolar organic solvent, the technique is straightforward for home chemists to exploit in order to refine their personal uranium stockpiles. The photo illustrates the supplies used in the following procedure: nitric acid, tri-n-butyl phosphate (from QualityBiological.com), Kleen-Strip 1-K kerosene (Home Depot), and 4.8 g of homemade uranyl oxide.
PUREX is the major chemical technique for recovering uranium from spent nuclear fuel. Based on the highly-selective extraction of uranyl nitrate from aqueous solution by tributyl phosphate (TBP) in a nonpolar organic solvent, the technique is straightforward for home chemists to exploit in order to refine their personal uranium stockpiles. The photo illustrates the supplies used in the following procedure: nitric acid, tri-n-butyl phosphate (from QualityBiological.com), Kleen-Strip 1-K kerosene (Home Depot), and 4.8 g of homemade uranyl oxide.
Caution: the PUREX procedure involves intimately contacting nitric acid with highly-flammable organic material! Work with small quantities. Concentrated acid will form explosive oils, so always dilute it to 6M or less. This discussion presupposes essential safety understanding of the chemicals and techniques involved.
_______________________________________________________________
Part I: Planning a Successful Extraction Scheme
.

Feedstock for the PUREX process is impure uranium dissolved in aqueous nitric acid. One question concerns the optimum acidity for the process, i.e., what quantity of acid results in the highest ratio of uranium concentration in the organic phase to that in the aqueous phase. This ratio [U]org/[U]aq = Kd is called the distribution coefficient. Kd is not constant, but depends on the concentrations of various dissolved species, on the temperature, and on other factors. At left is a plot taken from Marcus Y. (1961), relating log(Kd) to various acid concentrations in the aqueous phase, for some very low concentrations of uranium. The different curves represent different TBP concentrations in the organic phase.
The question about nitric acid concentration is answered conclusively here; the curves all peak between 5-6M. Higher acid concentrations are associated with a slow roll-off in Kd, while at lower concentrations it rapidly plunges even to the point that high aqueous concentrations of uranium are favored. Thus we should prepare an initial aqueous phase with 5-6M acid. The data also suggests that ~20-25 w/o TBP in kerosene are good values for the organic solution. I will plan a procedure around uranium oxide dissolved in excess 6M HNO3, and 20 w/o (0.66M) TBP / kerosene.
Actual values for Kd from Marcus’s plot are themselves not useful to us, because they relate to “tracer quantities” of uranium and we are interested in production quantities (more accurately, higher concentrations). To find quantitatively applicable data, we must look elsewhere.

At left is a McCabe-Thiele diagram relating [U]org in a TBP / kerosene phase to the equilibrium concentration [U]aq in an aqueous phase, for concentrations of uranium useful in production. In this particular data, from Stas J., Dahdouh A., et al. (2005), the concentrations were measured at room temperature, [HNO3]aq = 5.75M, and two different TBP solutions were used (0.726M, filled squares; 0.363M, circles). The 0.726M data is very applicable to our 6M HNO3 / 0.66M TBP preferences from above; as a tool for predicting extraction yields it should be accurate to within 10%.
We begin an extraction with a quantity of uranium dissolved in nitric acid, and a barren solution of TBP. During an extraction, the uranium concentration in the aqueous phase decreases by an amount Δ[U]aq. Simultaneously the uranium concentration in the organic phase rises by an amount Δ[U]org. Uranium is conserved (the amount lost from the aqueous phase must equal the amount gained in the organic), so Vaq×Δ[U]aq = Vorg×Δ[U]org. A line of slope Δ[U]org/Δ[U]aq = Vaq/Vorg = 1/Φ connects the initial state with the final equilibrium state on the black curve. Shown in red is the line for Φ = 1, [U]aq_1 = 0.34M. [U]org_1 is about 0.23M, meaning that on this first contact we can expect that about 68% of the uranium will have been extracted. If we save the aqueous layer and contact it again later (with bare organic), the blue line shows that we will have complexed essentially all the initially-present uranium with TBP. No more than two contacts will be needed to effectively process all the uranium.
The remaining component of PUREX is stripping pregnant organic phase with a barren aqueous phase in order to recover the uranium as purified uranyl nitrate. It’s evident that equilibrium favors uranium in the TBP at low concentrations (just look at the steep slope on the McCabe-Thiele diagram), so what can be done to move Kd in the direction of more aqueous uranium? Two easy things, evidently: raise the temperature, and reduce the nitric acid concentration. Data showing these effects is in the cited papers (and for nitric acid, the first figure above); unfortunately, these particular data are not quantitatively applicable to the present problem. I will use distilled water near the boiling point as the strippant, in as many contacts as are necessary to get the bright-yellow uranyl color out of the organic phase.
_______________________________________________________________
Part II: Doing The Experiment
.

The reactants. 69.5 g TBP was diluted to 400 ml with kerosene; 50 ml of the resulting solution was set aside for use in this extraction. 4.8 g of homemade UO3 were dissolved in 50 ml of 6M HNO3 to give a characteristic yellow solution that is approximately 0.34M in uranium and 5.4M in HNO3.
.
.
.


.
.
.
.
.
.
.
.
.
.
.
.
Both phases are placed in a separatory funnel and vigorously shaken together for about a minute. At left is before shaking, at right is after. The organic phase begins water-white and picks up a uranium-yellow coloration.
.

.
.
.
.
The aqueous (bottom) layer is now drained. While it still retains some coloration, it is not nearly as yellow as it was initially. The TBP / kerosene layer carries much of the uranium.
.
.
.
.
.
.


.
.
.
.
.
.
.
.
.
.
.
.
50 ml of hot distilled water is contacted with the uranium-loaded TBP / kerosene by shaking for about a minute, taking care to properly vent the kerosene vapors. The photo at left shows the colors before shaking; at right, after shaking and settling. Uranium is moving into the water.
The aqueous layer is drained and kept; the organic layer is washed again with a second 50-ml portion of water, which is also drained and added in with the first. I now repeat the entire procedure again on the original nitric acid solution—i.e. it has two contacts with the organic layer, and two more 50-ml portions of hot water strippant are collected.
.

.
.
.
.
.
The last water aliquot being collected, leaving behind a barren and nearly white organic layer.
.
.
.
.
.

.
.
.
.
.
.
.
.
.
.
The final state of the three solutions. The nitric acid solution containing impure uranium began bright yellow; now (diluted to 200 ml for a fair comparison) it is almost white. The four strippant aliquots combined are a nice yellow color. The organic solution is white. The original uranium has been transferred almost in its entirety to the strippant.
.
 Pure uranyl nitrate, UO2(NO3)2·6H2O, is recovered from the aqueous solution by evaporation of excess liquid in a microwave oven. This should be done outdoors (nitric acid vapors are evolved) with constant supervision; the melting point of uranyl nitrate is only 60°C and it is important to stop heating when excess water is gone and one begins decomposing the salt. The clue that this is happening is when little specks of orange uranyl oxide begin precipitating. Immediately remove the container from the oven and vigorously stir the molten salt with a stirring rod to prevent it from freezing into an intractable mass. A powder will result.
Pure uranyl nitrate, UO2(NO3)2·6H2O, is recovered from the aqueous solution by evaporation of excess liquid in a microwave oven. This should be done outdoors (nitric acid vapors are evolved) with constant supervision; the melting point of uranyl nitrate is only 60°C and it is important to stop heating when excess water is gone and one begins decomposing the salt. The clue that this is happening is when little specks of orange uranyl oxide begin precipitating. Immediately remove the container from the oven and vigorously stir the molten salt with a stirring rod to prevent it from freezing into an intractable mass. A powder will result.
My yield was 7.0 g, about 83% of the original uranium. Most of the losses can probably be chalked up to incomplete recovery of the nitrate after solidifying. Again, you must stir the living bejesus out of the salt as it freezes or it will stick on your glass like epoxy.

Well, what can I say? Superb! You shoud also share techniques of thorium extraction and purification as well!
Good work,
Andy
Very interesting, Carl.
Here’s a challenge for you.
You’ve got that AmBe source… if you were patient enough, I wonder how much plutonium one might be able to produce via neutron irradiation of natural uranium?
Of course, you’d have a little fission product radioactivity to contend with, as well as the more difficult chemistry involved with separating U from Pu in the solvent extraction.
You’d only be ever able to practically produce an infinitesimal, microscopic amount of Pu – but you’d only need an infinitesimal amount in order to prove that it’s present in the extraction solvent through alpha spectroscopy.
Show us that peak on the alpha spectrum at the right energy and you’ve achieved some kind of geek legend status forever 🙂
Hi Luke,
Neutron capture on uranium has been demonstrated at the “amateur” level, but with significantly more neutrons than are available from that AmBe source (specifically, Farnsworth fusors producing neutrons by DD fusion). Jon Rosenstiel activated U-238 to form U-239, which is a precursor to Pu-239. The radioactive decay of U-239 can be detected by high-resolution gamma spectroscopy:
http://www.fusor.net/board/view.php?bn=fusor_neutrons&key=1192914434
I don’t have an operative HPGe detector, but I have detected fission products in natural uranium after exposure to fusor neutrons, using scintillation detectors (details and link on my Farnsworth fusor post).
-Carl
Wow! a brilliant post…clear and concise..
Would the Purex process work for extracting other element, such as, Br, Ca, Cl, Co, Fe, K, SiO2, SO4, Ti, and Zr? I am trying to get all of these components in a Kerosene/TBP matrix.
Not only PUREX process, any extraction process involving an organic solvent is able to extract the solutes which can form organic soluble, coordinated complexes. For that the metal solute should have an empty electron orbital, which are not available with alkali and alkaline earth metals. If this condition is satisfied, it is possible.
YOU ARE ALL NERDS!!!
I’m so confused at how you can do all this and not have any trouble whatsoever with the regulatory agencies…
10 CFR § 40.13 Unimportant quantities of source material. (Redacted for length by Carl Willis. We can all find the regulation text online if interested)
Awesome explanation!!!
150gram
Very interesting and useful! Iam trying to develop my own homogeneous reactor so this tech will help to get clear fuel from ore.